| The deficiency of N-linked sugar chain transfer is detected in congenital carbohydrate-deficient glycoprotein syndrome (later, referedas congenital disorders of glycosylation, CDG) (1) and in posterior alcohol abuse (2). On the other hand, the hyperglycosylation is reported in Alzheimer's disease (3).
CDG, an autosomal recessive congenital disorder first reported in 1980, is characterized by multisystemic features including cerebellar hypoplasia/atrophy, hepatopathy and polyneuropathy. Biochemical studies revealed that various serum glycoproteins show higher isoelectric points by the deletion of acidic sugar chains. CDGS is grouped into two types; type I is an N-linked sugar chain transfer deficiency, type II is a deficiency of N-acetylglucosaminyltransferase II in the biosynthetic pathway at Golgi apparatus.
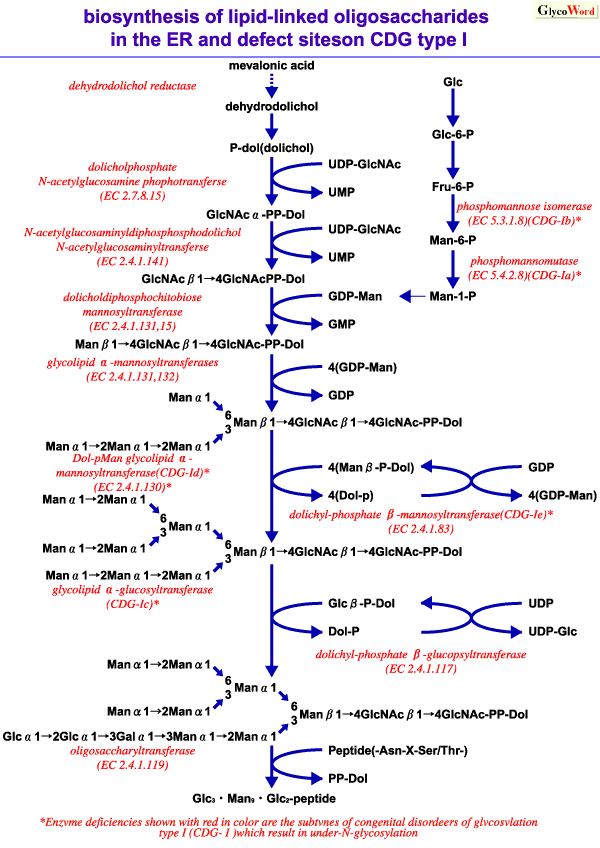
CDGS type I is further classified in to several subtypes by deficient enzymes; phosphomannomutase deficiency (type Ia)(4), which is most frequent, phosphomannose isomerase deficiency (Type Ib)(5), glucosyltransferase deficiency and dehydrodolichol reductase deficiency are reported. In all subtypes of type I, the levels of Glc3Man9GlcNAc2-PP-dolichol decrease, and as a result, the transfer efficiency of oligosaccharides to polypeptides decreases. Further new oligosaccharide transfer deficiencies by defects of the other enzymes might be discovered, because the biosynthetic process of dolichol-linked oligosaccharide intermediate consists of multi-steps. Up to the present, the transfer deficiency of N-linked glycan has been reported on serum glycoproteins such as transferrin and antithrombin III, beta-trace protein from cerebrospinal fluid, and erythrocyte membrane glycoproteins in patients with CDG type I, and such transfer deficiency should occur in most tissues because of its broad clinical symptoms. It has been reported that loss of N-linked sugar chains affects the biological function of glycohormones, cell-surface expression of receptors, secretion of glycoproteins and intracellular transport of lysosomal enzymes. Although the symptoms of CDG type I are crucial in early infancy, they gradually improve, and patients even older than 50 years of age are reported. No definitive treatment is currently available for CDG. However, oral supplementation with mannose improved the biochemical abnormalities in the deficiency of phosphomannose isomerase (CDG Type Ib) so that symptoms were relatively slight.
N-glycosylation defects of serum glycoproteins are also detected in individuals with chronic alcohol consumption. In particular, carbohydrate-deficient transferrin (CDT) is a specific marker for alcohol abuse, and disappears with abstinence, but the mechanism of the aglycosylation has not yet been elucidated.
Hyperglycosylation, that is, unusual transfer of sugar chain to potential N-glycosylation site (-Asn-X-Ser or Thr-) to which the sugar chain cannot usually bind by conformational hinderance, has been reported in tau protein of patients with Alzheimer's disease. This phenomenon may occur at an abnormal arrangement of membrane or by dysfunction of chaperones.
| |
|
| References | (1) | Yamashita, K, Ideo, H, Ohkura, T, Fukushima, K, Yuasa, I, Ohno, K, Takeshita, K : Sugar chains of serum transferrin of patients with carbohydrate deficient glycoprotein syndrome: Evidence of asparagine- N-linked oligosaccharide transfer deficiency. J. Biol. Chem. 268, 5783-5789, 1993 |
| (2) | Landberg, E, Pahlsson, P, Lundblad, A, Arnetorp, A, Jeppsson, JO : Carbohydrate composition of serum transferrin isoforms from patients with high alcohol consumption. Biochem. Biophys. Res. Commun. 210, 267-274, 1995 |
| (3) | Wang, J-Z, Grundke-Iqbal, I,Iqbal, K : Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer's disease. Nature Medicine, 2:871-875, 1996 |
|
(4) |
Matthijs, G. et al. : Mutations in PMM2, a phosphomannomutase geneon chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome(Jaeken syndrome), Nature Genetics, 16, 88-92, 1997 |
|
(5) |
Niehues, R. et al. : Carbohydrate-deficient glycoprotein syndrome typelb. Phosphomannose isomerase deficiency and mannose therapy, J. Clin. Invest.,101, 1414-1420, 1998 |
|
(6) |
Imbach, T. et al. : A mutation in the human ortholog of the Saccharomycescerevisiae ALG6 gene causes carbohydrate-deficient glycoprotein syndrometype-Ic, Proc.Natl. Acad. Sci. USA, 96, 6982-6987, 1999 |
|
(7) |
Kaner, C. et al. : Carbohydratedeficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man5GlcNAc2-PP-dolichylmannosyltransferase, EMBO J., 18, 6816-6822, 1999 |
|
(8) |
Imbach T, Schenk B, Schollen E, Burda P, Stutz A, Gr]unewald S, BailieNM, King MD, Jaeken J, Matthijs G, Berger EG, Aebi M, Hennet T. Deficiencyof dolichol-phosphate-mannose synthase-1 causes congenital disorder ofglycosylation type Ie.J Clin Invest.,105, 233-9, 2000 |
| | |
| |