 |
Impairment of keratan sulfate proteoglycan (KS-PG) synthesis is categorized into two groups: impaired production of core protein and KS side chain of KS-PG. There are three KS-PG core proteins in the human cornea, lumican, keratocan, and mimecan, all of which belong to a group of small leucine-rich repeat proteins. Mutations on a gene for keratocan (KERA) cause an autosomal recessive eye disease called cornea plana, which is characterized by reduced corneal curvature leading hyperopia (1). Pathogenesis of this disease is a functional defect of keratocan protein rather than a lack of KS synthesis, since other KS-PGs are produced in the tissue. Therefore, this disease is not discussed in this section.
In human cornea, four Golgi-residing enzymes (β4GalT4, β3GnT7, CHST1 and CHST6) are involved in KS synthesis (see “Biosynthesis of keratan sulfate and its sulfation steps”). Among them, only CHST6 has been reported to be involved in human genetic disease. Loss of function mutation of CHST6 causes macular corneal dystrophy (MCD), an autosomal recessive disease that the patients develop punctate deposits in their corneal stroma. The deposits begin to appear at the age of 5 to 9, and gradually spread throughout the cornea as age increases. Eventually the cornea becomes completely cloud, and the patients requite to have corneal transplantation. Large families of MCD have been reported in Iceland.
Symptoms of MCD are limited to the cornea in human. Quantock et al. reported that MCD cornea is thinner than normal cornea due to reduced interspacing of collagen fibrils in the corneal stroma (3). Biochemical analysis revealed absence of immunoreactive sulfated KS in the serum of MCD patients (4). After this report, several groups analyzed serum and corneal KS level using anti-KS antibody 5D4, and classified MCD patients into three groups: 1) type I, in which immunoreactive KS is not detected in the serum or the cornea; 2) type IA, which is almost identical to type I, but immunoreactive KS is detected in the corneal stromal cells (keratocytes); 3) type II, in which KS is detected both in the serum and the cornea (5). Since the most MCD cases are classified as type I, in which immunoreactive sulfated KS is absent in the extracellular matrix of the corneal stroma, abnormal KS biosynthesis is thought to be the cause of MCD. Using radiolabel tracing, Nakazawa et al. analyzed glycoproteins produced from organ-cultured MCD corneas and found the corneal cells produced unsulfated KS-PG, indicating that sulfation step of KS biosynthesis is impaired in the MCD cornea (6).
The critical region for the causative mutation of MCD is located on chromosome 16q22-23 (7), and the gene was identified by searching a carbohydrate sulfotransferase gene on the critical region. CHST5 and CHST6 are located in that region of human genome, but all the causative mutations of MCD were on CHST6. These two genes, CHST5 and CHST6, are present in primate genomes, but only one of the genes is present in the genome of other animal species (8). CHST5 and CHST6 exist side by side in human genome only 30 kbp apart, and the entire genomic sequences are homologous to each other, indicating these two genes are created by gene duplication. CHST6 encodes N-acetylglucosamine (GlcNAc) 6-O-sulfotransferase CHST6 (also called as corneal GlcNAc 6ST/GST4β/GlcNAc6ST-5). In genome of MCD type I patients, deletion, frameshift and missense mutations, all of which result in inactivation of the enzyme, were found as homozygotes or compound heterozygotes. On the other hands, Akama et al. analyzed genomic sequence upstream of CHST6 protein coding region, which is presumed to be the promoter region of CHST6, and found deletion and replacement mutations that may have resulted from homologous recombination between CHST5 and CHST6. They also examined expression of CHST6 in MCD type II cornea by in situ hybridization, and found lack of expression in corneal epithelial cells. These results concluded that lack of CHST6 expression in the corneal cells leads to MCD type II phenotype (9). Because large amount of long KS chain must be produced in the cornea, inactivation of CHST6 enzyme (MCD type I) and lack of CHST6 expression (MCD type II) may impact on glycan production in the tissue.
Defect of CHST6 activity results in production of abnormal KS that have no sulfation on 6-O position of GlcNAc. Since CHST1 (also known as KS galactose 6-O sulfotransferase) enzyme, which is responsible for galactose sulfation of KS, prefers GlcNAc-6-O-sulfated glycans as its substrates (10), inactive CHST6 not only results in lack of GlcNAc sulfation but also reduces sulfation on galactose, leading production of poly-N-acetyllactosamine (polyLacNAc) instead of KS. PolyLacNAc has distinct characteristics from KS in that it is more hydrophobic and less water solubility than KS due to lack of electric charge on the carbohydrate chains. In the corneal stroma, collagen fibrills are surrounded by KS-PG, and interfibrillar spaces are filled with hydrophilic macromolecules, including KS-PG, and water molecules, maintaining corneal transparency. Therefore, reduction of KS-PG production leads to shortening of collagen fibril interval, which results in thinning of corneal stromal thickness, and production of hydrophobic polyLacNAc may cause aggregation and precipitation of the glycan in the cornea, resulting in clouding of the tissue.
Although CHST5 has sulfotransferase activity in vitro, that enzyme cannot produce KS (11, 12). MCD type II patients with homozygous mutation lacking entire CHST5 showed no extra phenotype other than MCD (9). To date, the biological function of CHST5 is unknown.
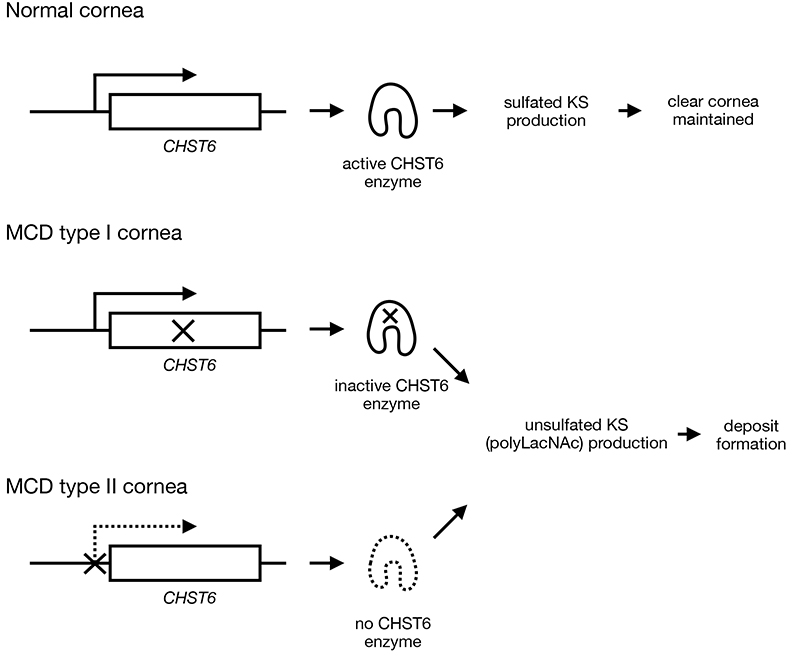
Fig. 1. Pathogenesis of MCD
In the corneal stroma, KS-PG binds to collagen fibrils to arrange the fibrils in order; it also fills the interfibrillar space, together with water and other hydrophilic macromolecules, to maintain high transparency. In MCD patients with loss-of-function mutations on CHST6 (MCD type I), which lead to inactive CHST6 production, or with mutations on gene-regulatory region of CHST6 (MCD type II), which result in lack of CHST6 enzyme production, unsulfated KS (polyLacNAc), is produced and secreted from the corneal cells. Since polyLacNAc is more hydrophobic than KS, it aggregates and precipitates in the corneal stroma, causing corneal opacity.
|
Tomoya Akama
(Department of Pharmacology, Kansai Medical University)
| References |
| (1) |
Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, Kivelä T, Kucherlapati R, Forsius H, de la Chapelle A: Mutations in KERA, encoding keratocan, cause cornea plana. Nat. Genet. 25, 91-95, 2000 |
| (2) |
Singh S, Das S, Kannabiran C, Jakati S, Chaurasia S: Macular corneal dystrophy: An updated review. Curr. Eye Res. 46, 765-770, 2021 |
| (3) |
Quantock AJ, Meek KM, Ridgway AE, Bron AJ, Thonar EJ: Macular corneal dystrophy: reduction in both corneal thickness and collagen interfibrillar spacing. Curr. Eye Res. 9, 393-398, 1990 |
| (4) |
Thonar EJ, Meyer RF, Dennis RF, Lenz ME, Maldonado B, Hassell JR, Hewitt AT, Stark WJ Jr, Stock EL, Kuettner KE, Klintworth GK: Absence of normal keratan sulfate in the blood of patients with macular corneal dystrophy. Am. J. Ophthalmol. 102, 561-569, 1986 |
| (5) |
Klintworth GK, Oshima E, al-Rajhi A, al-Saif A, Thonar EJ, Karcioglu ZA: Macular corneal dystrophy in Saudi Arabia: a study of 56 cases and recognition of a new immunophenotype. Am. J. Ophthalmol. 124, 9-18, 1997 |
| (6) |
Nakazawa K, Hassell JR, Hascall VC, Lohmander LS, Newsome DA, Krachmer J: Defective processing of keratan sulfate in macular corneal dystrophy. J. Biol. Chem. 259, 13751-13757, 1984 |
| (7) |
Vance JM, Jonasson F, Lennon F, Sarrica J, Damji KF, Stauffer J, Pericak-Vance MA, Klintworth GK: Linkage of a gene for macular corneal dystrophy to chromosome 16. Am. J. Hum. Genet. 58, 757-762, 1996 |
| (8) |
Akama TO, Fukuda MN: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5 and 6 (CHST5,6). In: Handbook of Glycosyltransferases and Related Genes. Taniguchi N, Honke K, Fukuda M, Narimatsu H, Yamaguchi Y, Angata T (editors), Springer Tokyo, Tokyo, Japan, 1005-1014, 2014 |
| (9) |
Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T, Dota A, Kawasaki S, Inoue Y, Maeda N, Yamamoto S, Fujiwara T, Thonar EJ, Shimomura Y, Kinoshita S, Tanigami A, Fukuda MN: Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat. Genet. 26, 237-241, 2000 |
| (10) |
Fukuta M, Inazawa J, Torii T, Tsuzuki K, Shimada E, Habuchi O: Molecular cloning and characterization of human keratan sulfate Gal-6-sulfotransferase. J. Biol. Chem. 272, 32321-32328, 1997 |
| (11) |
Akama TO, Nakayama J, Nishida K, Hiraoka N, Suzuki M, McAuliffe J, Hindsgaul O, Fukuda M, Fukuda MN: Human corneal GlcNAc 6-O-sulfotransferase and mouse intestinal GlcNAc 6-O-sulfotransferase both produce keratan sulfate. J. Biol. Chem. 276, 16271-16278, 2001 |
| (12) |
Akama TO, Misra AK, Hindsgaul O, Fukuda MN: Enzymatic synthesis in vitro of the disulfated disaccharide unit of corneal keratan sulfate. J. Biol. Chem. 277, 42505-42513, 2002 |
Oct. 12, 2023
|
|---|





